LEARNING OBJECTIVES
2. Understand the pathophysiology of stable and unstable coronary plaques.
3. Be able to identify several mechanisms by which unstable angina can occur.
4. Understand mechanisms of acute myocardial infarction.
5. Understand pathophysiology of various complications of acute MI, including right ventricular infarction, congestive heart failure, heart block, acute myocardial rupture, acute mitral regurgitation, and cardiogenic shock.
6. Understand mechanisms by which the various ischemic syndromes (stable and unstable angina, and acute MI) can be treated medically and with interventional therapies.
7. Be able to identify the major risk factors for
development of coronary artery disease.
What is ischemia?
Imbalance between supply and demand resulting in
one of many clinical syndromes.
| SYNDROME | DESCRIPTION |
| Ischemic heart disease | Condition in which an imbalance between myocardial oxygen supply and demand results in myocardial hypoxia and accumulation of waste metabolites; most often due to atherosclerotic disease of the coronary arteries ("coronary artery disease") |
| Angina pectoris | Uncomfortable sensation in the chest or neighboring anatomic structures produced by myocardial ischemia |
| Stable angina | Chronic pattern of transient angina pectoris, precipitated by physical activity or emotional upset, relieved by rest within a few minutes; episodes often associated with temporary depression of the ST segment, but permanent myocardial damage does not result |
| Variant angina | Typical anginal discomfort, usually at rest, which develops because of coronary artery spasm, rather than an increase of myocardial oxygen demand; episodes often associated with transient shifts of the ST segment (usually ST elevation) |
| Unstable angina | Pattern of increased frequency and duration of angina episodes, produced by less exertion, or at rest; high frequency of progression to myocardial infarction if untreated |
| Silent ischemia | Asymptomatic episodes of myocardial ischemia; can be detected by EKG and other laboratory techniques |
| Myocardial infarction (See Chapter 7) | Region of myocardial necrosis usually due to prolonged cessation of blood supply; most often results from acute thrombus at site of coronary atherosclerotic stenosis; may be first clinical manifestation of ischemic heart disease, or there may be a history of angina pectoris |
Determinants of myocardial blood supply and consumption (demand)
Supply:
1. Coronary blood flow
2. Oxygen saturation of blood
3. Hemoglobin content of blood
Coronary blood flow is proportional to coronary perfusion pressure divided by coronary vascular resistance.
Coronary perfusion pressure is determined primarily by the aortic diastolic pressure and can be reduced abruptly beyond the site of high grade (> 70%) coronary artery stenoses.
Coronary vascular resistance is determined by external compressive forces as well as metabolic, endothelial, and neural factors which regulate intrinsic coronary tone via auto- regulation.
Oxygen saturation of the blood can be reduced by interference with the alveolar-capillary interface, as can occur in many lung diseases. Oxygen saturation could also be reduced by a right-to-left shunt causing admixture of venous and arterial blood.
Hemoglobin content of the blood is reduced in any
condition causing anemia.
Demand:
2. Heart rate
3. Myocardial contractility
|
|
( P x r / 2h ) |
|
|
· external compression · intrinsic regulation - local metabolites - endothelial factors - neural innervation |
Heart rate | |
| 02-carrying capacity |
Contractility |
THE CORONARY ATHEROSCLEROTIC PLAQUE
Perhaps the most common mechanism of coronary ischemia is the development of coronary atherosclerotic plaque resulting in reduced myocardial oxygen supply. Atherosclerotic plaques most likely develop at sites of fatty streaks, in areas where there are accumulations of "foam cells" which are filled with lipid-laden macrophages. Plaques may be either "soft" or "hard", depending on the ratio of lipid-laden foam cells to fibrous tissue within the plaque. Recent research suggests that softer, lipid-laden plaques may be more prone to plaque rupture resulting in unstable angina or acute myocardial infarction. A schematic diagram of "plaque evolution" is presented below.
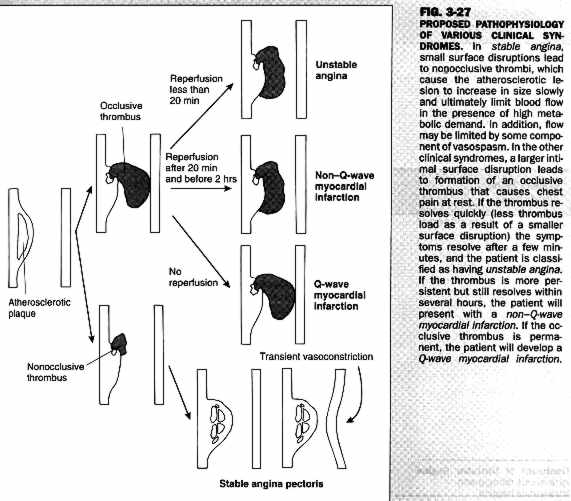
Risk factors for the development of atherosclerotic plaques include:
2. Cigarette smoking
3. Hypertension
4. Diabetes mellitus
5. Family history of premature coronary artery
disease
ACUTE MYOCARDIAL INFARCTION
An abrupt disruption of the coronary artery plaque, leading to occlusive thrombus formation is the most common cause of acute myocardial infarction. This initiates an "ischemic cascade", which progresses as outlined below:
2. Decreased systolic function (usually localized)
3. Ischemic electrocardiographic changes
4. Clinical symptoms of chest discomfort or other "anginal equivalents"
5. Release of breakdown products due to cell death, i.e. CPK, LDH, troponin-i enzymes.
| TIME | EVENT |
| Early changes
|
|
| 1 - 2min | ATP levels fall; cessation of contractility |
| 10 min | 50% depletion of ATP; cellular edema, decreased membrane potential and susceptibility to arrhythmias |
| 20 - 24 min | Irreversible cell injury |
| 1 - 3 hours | Wavy myofibers |
| 4 - 12 hours | Hemorrhage, edema, PMN infiltration |
| 18 - 24 hours | Coagulation necrosis (pyknotic nuclei with eosinophilic
cytoplasm), edema |
| 2-4 days | Total coagulation necrosis (no nuclei or striations, rimmed by hyperemic tissue); monocytes appear |
| Late Changes
|
|
| 5 - 7 days | Yellow-softening from resorption of dead tissue by macrophages |
| 7 days + | Ventricular remodeling |
| 7 weeks | Fibrosis and scarring complete |
Clinical presentation of acute MI
Usually located in the center of the chest over an area the size of a fist rather than in a localized point. There is often radiation of discomfort to either or both upper arms or shoulders, sometimes to the neck and into the jaw or into the back. There is often accompanying diaphoresis (sweatiness), shortness of breath, and nausea or vomiting.
The physical exam may be entirely unremarkable but may demonstrate an S4 gallop, occasionally an S3 gallop if myocardial infarction is large and leads to significant left ventricular dysfunction. Occasionally a dyskinetic apical impulse may be present if an extensive antero-apical MI occurs. A systolic murmur may occur with acute mitral regurgitation or ventricular/septal defect. Jugular venous distention (JVD) may be present in the setting of right ventricular infarct or when there is significant biventricular heart failure. Pulmonary rales may be present if left ventricular failure is present.
Electrocardiographic changes include ST segment elevation followed ultimately by T-wave inversion and evolution of Q-waves over a time course of 24 to 48 hours.
The location of the myocardial infarction is dependent on the site of coronary thrombosis as are the potential complications which may follow. Coronary artery anatomy is reviewed below:

Of the three major coronary arteries (left anterior descending, circumflex, and right) the left anterior descending supplies proportionately the most myocardium, perhaps 40% on average. Occlusion of the proximal LAD, therefore, may cause a large area of myocardial necrosis and this may lead to severe left ventricular impairment and congestive heart failure.
Since a large proportion of the intraventricular septum is supplied by the LAD, conduction system structures which course through the septum (the bundle of His and the His-Purkinji system) may be injured. This may result in heart block (LBBB, RBB, Mobitz II A-V block, or complete heart block) which may be irreversible and require permanent pacemaker placement. If the anterior infarct is large and involves the entire apex of the heart, intraventricular thrombus may occur and lead to embolization.
Occlusion of the right coronary artery
usually causes less impairment to left ventricular function. The right
ventricle, however, is supplied primarily by the right coronary artery
and right ventricular infarction may occur (see P. 9.) The S/A nodal artery
is supplied 70% of the time from the right coronary artery and therefore
sinus node dysfunction and slowing may occur. The AV nodal artery arises
from the posterior descending branch, which is a branch of the right coronary
artery in 85% of individuals. Therefore, transient heart block at the level
of the A-V node may occur (1st degree, Mobitz I, or 3rd
degree A-V block), which is usually reversible and does not require permanent
pacemaker implantation. Finally, the posterior descending artery is the
primary source of blood to the posterior papillary muscle which may be
prone to muscular rupture in the setting of a large inferoposterior MI.
Please refer to
Lecture 5: Complications of AMI
TREATMENT OF ISCHEMIC HEART DISEASE
The treatment of the patient with ischemic coronary artery disease depends in large part on the presenting clinical syndrome. Patients with stable and unstable angina are treated initially with antiplatelet therapy (aspirin) to reduce the likelihood of clot formation and therefore to reduce the chance of acute MI. Intravenous heparin is also a mainstay of treatment for unstable angina, and works by inhibiting further clot formation. Other, newer anti-platelet agents include several intravenously administered potent inhibitors of platelet aggregation called the 2b,3a inhibitors, which are also reserved for unstable anginal syndromes.
Beta-blockers can be administered orally or intravenously to decrease myocardial oxygen demand by reducing heart rate and contractility and, by reduction of blood pressure, wall tension.
Nitrates are effective in reducing venous return to the heart (pre-load) thereby reducing wall tension. Another effect of organic nitrates is to cause a direct vasodilatation of the coronary arteries, thereby increasing coronary perfusion.
Calcium channel blockers are a relatively large class of agents which act by different mechanisms. Some of them act more through peripheral vasodilatation and reduction of blood pressure, i.e. wall stress, while others act through negative chronotropic effects, thereby reducing heart rate and again reducing oxygen demand. All of them cause some coronary vasodilatation to lesser or greater extents, resulting in increased coronary blood flow.
Thrombolytic therapy remains the most important modality for the treatment of acute myocardial infarction and, in the absence of contra-indications, is recommended for all patients presenting within the first several hours of myocardial infarction. Thrombolytic therapy is usually combined with longer-acting anti-coagulants including heparin and anti-platelet agents. In addition, intravenous nitroglycerin and often ß-blockers are added to the regimen for acute myocardial infarction.
Nonmedical modalities will be discussed briefly and include:
2) Directional coronary atherectomy (DCA)
3) Coronary artery bypass grafting (CABG)