The maps code is started using the command
where the option -d indicates that all default values of the input parameters should be used, which is what most users
will ever need. (The optional parameters can be displayed by typing maps by itself and further help is available via the command
maps -h.) The trailing & character cause the command
to execute in “background” mode. In this fashion, maps can continuously be on the lookout, responding to various “signals”, while
the user performs other tasks. (The ongoing discussion assumes that the code is run under a UNIX
environment within a shell such as sh, csh, tcsh or bash.)
The process of constructing a cluster expansion from first-principles calculations can be summarized as follows.
- Determine the parameters of the first-principles code that provide the desired accuracy.
- Let maps refine the cluster expansion.
- Decide when the cluster expansion is sufficiently accurate.
Typically, one calibrates the accuracy of the first-principles calculations using the “pure”5.1 structures of the alloy system of interest.
To generate the two “pure” structures, type
This creates a file called ready which tells maps that you are ready to calculate the energy of a structure.
Within 10 seconds, maps replies with
| Finding best structure... |
| done!
|
maps has just created a directory called 0 and, within it, a file called str.out that contains the geometry of one of the two “pure” structures.
If you type touch ready once more, the other “pure” structure is written to 1/str.out.
You now need to launch the first-principles code to calculate the energy of each structure.
Type
to go into the directory of the first structure. Assuming that your first-principles code is called xxxx, type
After this command has successfully terminated, display the energy of that structure and
go back to the initial directory
and edit the file defining the first-principles code parameters
so that the precision of the calculation is increased (e.g. increase the
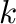 -point density or the cut-off of the plane-wave energy). Then you rerun
the calculations to check by how much the calculated energy has changed:
-point density or the cut-off of the plane-wave energy). Then you rerun
the calculations to check by how much the calculated energy has changed:
| cd 0 |
| runstruct_xxxx & |
| cat energy (After the calculations are completed) |
| cd ..
|
This process is repeated until the user is satisfied with the precision of the calculation
(that is, if the energy has become insensitive to changes in the input parameters within
the desired accuracy).5.2A similar study should also be performed for the other “pure” structure (labeled structure
1) and, if one is really concerned with precision, for a few structures with
intermediate concentrations.
Once the appropriate ab initio code parameters have been determined, the fully automated
process can begin. From within the directory where maps was started,
type
to start the job manager that will monitor the load on your local network of workstations and ask maps
to generate new structures (i.e. atomic arrangements) whenever a processor becomes available.
Note that the first time the command is run, instructions will appear on screen
that explain how to configure the job dispatching system in accordance to your local
computing environment. Once this configuration is complete, the above command should
be invoked in the background by appending a “&” to it.
avdw@alum.mit.edu Wed, Dec 6, 2023 12:55:16 PM